
学术交流
过氧化氢(H₂O₂)作为一种仅含氧和氢的清洁氧化剂,在废水处理、纸浆漂白及化学合成等众多工业过程中扮演着不可替代的角色。然而,目前工业上几乎全部依赖蒽醌法进行生产,该工艺不仅能耗密集、依赖贵金属催化剂,还会伴随有机副产物的生成,带来环境负担。近年来,利用电催化两电子氧还原反应(2e⁻ ORR)在温和条件下直接合成H2O2被视为绿色替代路线,但该路线长期受困于催化剂的活性与选择性难以兼得,尤其是p区金属(如Bi)基催化剂面临着选择性调控机制不清和质子供给不足的双重瓶颈。
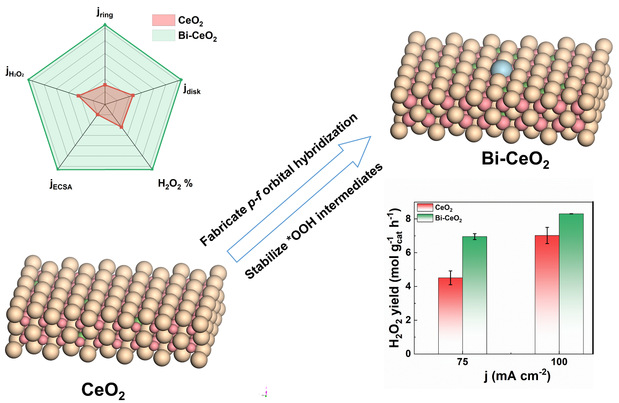
图1 不对称Bi-Ce双位点催化剂通过p-f轨道杂化协同促进H2O2电合成机理示意图
针对上述难题,高嵩博士团队通过原子级构型设计,成功构建了Bi-CeO2不对称双金属位点催化剂,并首次揭示了Bi 6p与Ce 4f轨道杂化(p-f orbital hybridization)协同促进H2O2电合成的全新机制。研究发现,Bi3+引入CeO2晶格后,不仅通过离子半径失配(Bi3+ 1.08 Å vs Ce4+ 0.92 Å)诱导晶格应变与氧空位富集,更重要的是,Bi与相邻Ce位点之间形成了p-f轨道耦合,实现了电荷的定向重新分布,并促使表面暴露的Ce3+位点成为电子富集中心。该双位点结构能够同时优化氧气吸附构型、高效解离水分子提供质子,并选择性地稳定关键中间体*OOH,从而打破了传统催化剂在活性与选择性之间的线性标度关系。
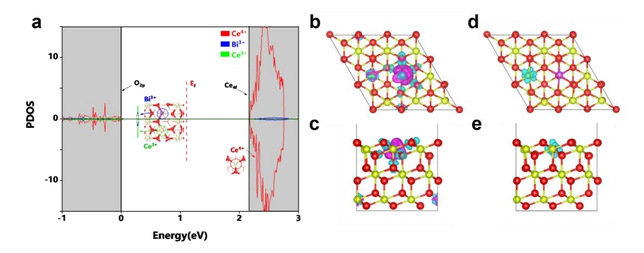
图2 DFT理论计算揭示Bi-CeO2中p-f轨道杂化及电荷局域化分布
实验结果表明,最优的1.6 wt.% Bi-CeO2催化剂在碱性条件下实现了93%的H2O2选择性。在气体扩散电极(GDE)流动池测试中,该催化剂在100 mA cm⁻²的工业级电流密度下,达到了8.3 mol gcat⁻¹ h⁻¹的H2O2产率,并展现出优异的长时间运行稳定性。原位红外光谱(ATR-SEIRAS)检测到了关键的*OOH中间体信号,而密度泛函理论(DFT)计算进一步证实,Bi与Ce之间的p-f轨道杂化导致电荷在Bi-Ov-Ce局域构型中重新分布,显著降低了氧空位形成能(仅0.62 eV),并提升了界面质子转移动力学(动力学同位素效应KIE接近1)。这一系列表征与计算共同揭示了p-f轨道杂化驱动下的“电子-质子”协同调控机制,正是该机制赋予了Bi-CeO2催化剂优异的选择性、高产率与长效稳定性,为非贵金属电催化剂设计提供了新的理论依据。
该研究成果以论文形式发表于《Journal of Catalysis》期刊,题目为“Asymmetrical Bi and Ce Diatomic Sites: p-f Orbital Hybridization Synergistically Boosts Hydrogen Peroxide Electrosynthesis via Oxygen Reduction”。高嵩博士为该论文的第一作者。《Journal of Catalysis》是国际催化领域顶级期刊,1962 年创刊,专注刊发多相、均相催化领域具备重大基础创新的系统性原创研究。
该研究得到了国家自然科学基金项目的资助。
原文链接:https://doi.org/10.1016/j.jcat.2026.116989






